Болезнь Фабри
Типичные проявления болезни Фабри:
потеря зрения, глазные болезни (изменение роговицы, катаракта);
приступы боли в руках и ногах;
чрезмерная потливость, поражения кожи (ангиокератомы);
сердечно-сосудистые расстройства;
боли в животе, тошнота, диарея.
Могут нарушиться работа почек и развиться артериальная гипертензия почечного происхождения, которая провоцирует сердечные расстройства и вызывает поражение артерий головного мозга. В некоторых случаях имеет место деформация суставов, некроз костей, задержка роста и полового созревания.
Симптомы болезни Фабри проявляются в первые 10 лет жизни. В основном, пациенты жалуются на боль в руках и ногах, которая проходит через несколько дней. У женщин признаки заболевания выражены не так сильно и появляются позже.
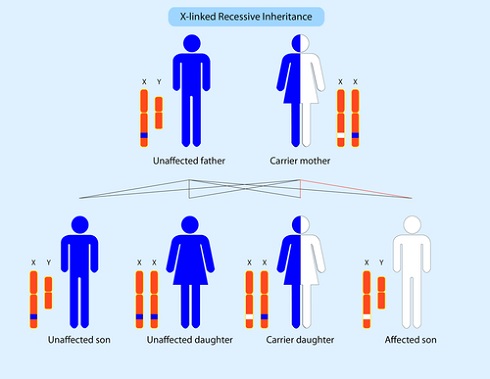
Болезнь Фабри — генетическое заболевание. Дефицит фермента альфа-G4-галактозидазы приводит к накоплению гликолипидов в цитоплазме и лизосомах клеток разных органов: в эндотелиальных клетках лимфатических и кровеносных сосудов, клетках гладкой мышечной ткани кровеносных сосудов, в клетках центральной и периферической нервной системы. Фермент может полностью отсутствовать или вырабатываться в очень малом количестве. Заболевание наследуется по Х-сцепленному рецессивному типу.
Если болезнь Фабри не лечить, продолжительность жизни больного может сократиться примерно на 20 лет. Заболевание приводит к повреждению почек (почечная недостаточность), сердца (недостаточность сердечных клапанов), и мозга (ишемический инсульт). Смерть, чаще всего, наступает в возрасте около 40 лет из-за уремии или сосудистых поражений сердца и головного мозга.
Болезнь Фабри диагностируют довольно поздно, в среднем — через 8 лет после появления первых признаков: глазной патологии, изменений на коже и слизистых оболочках, парестезии, расстройств терморегуляции тела. Подтверждают диагноз лабораторные исследования. Повышенная активность альфа-галактозидазы в лейкоцитах, плазме крови, биоптатах тканей, повышенный уровень сфингогликолипидов в моче свидетельствуют о наличии болезни Фабри.

Лечение
Прогрессирование заболевания может быть замедленно с помощью заместительной терапии. Пациенту вводят генетически синтезированный фермент альфа-галактозидазы. Так как период полураспада фермента короткий, инъекции необходимо делать раз в две недели на протяжении всей жизни. Однако фермент может вызвать побочные эффекты — температура, тошнота, озноб, аллергическая реакция. Поэтому на начальном этапе необходимо осуществлять инъекции под наблюдением врача, в дальнейшем это можно делать самостоятельно.
В дополнение к этому проводят симптоматическую терапию. Назначают обезболивающие, а в сложных случаях может потребоваться пересадка почки. Особенно успешно лечение проходит, если болезнь диагностирована в детском возрасте. В этом случае возможно предотвратить повреждение большинства органов.
Профилактика
Так как болезнь Фабри — генетическое заболевание, профилактических мер для него не существует. Диагностировать данное заболевание можно уже на 15 неделе беременности. Если родители страдают от болезни Фабри, необходимо как можно раньше проверить ребёнка. Ранняя диагностика и своевременное лечение помогут замедлить течение болезни и предотвратить возникновения тяжёлых осложнений.